Agendar
Exames, vacinas e mais- Home
- Quando a causa da anemia é genética
Quando a causa da anemia é genética
Apesar de haver centenas de variantes, apenas alguns defeitos causam síndrome clínica. Veja quais são e como rastreá-losAs hemoglobinopatias compõem um grupo heterogêneo de doenças genéticas decorrentes de alterações moleculares nos genes das globinas, os quais respondem pela síntese de hemoglobina (Hb). Estima-se que 7% da população mundial seja portadora de algum tipo dessas entidades hematológicas.
Atualmente, são descritas por volta de 1.700 variantes estruturais (Globin Gene Server – Nature Genetics: doi:10.1038/ng.785), porém poucas estão relacionadas a manifestações clínicas e alterações hematológicas. Quando a mutação altera a estabilidade da molécula ou suas propriedades funcionais, como a afinidade pelo oxigênio ou a solubilidade, o defeito genético pode se associar à síndrome clínica.
Entre as mais frequentes hemoglobinopatias estão a anemia falciforme, a hemoglobinopatia C e as anemias hemolíticas decorrentes das hemoglobinas instáveis. Em muitos casos, as mutações são determinadas por distribuições étnicas e geográficas, fundamentando os programas de controle de tais alterações e o aconselhamento genético.
No Brasil, as anormalidades mais relevantes, devido à sua prevalência, incluem a Hb S, a Hb C, a Hb D-Los Angeles e as talassemias, que são detalhadas a seguir.
Talassemias
São relativamente comuns em diversas populações do Mediterrâneo (italianos, portugueses, espanhóis e libaneses), que imigraram para o Brasil em grandes contingentes, especialmente no Sul e no Sudeste. Sua prevalência média é de cerca de 1% na população geral, podendo se elevar em comunidades isoladas. Pode se apresentar como betatalassemia ou alfatalassemia.
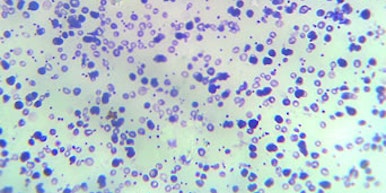
 Caso de betatalassemia demonstrado por microscopia.
Caso de betatalassemia demonstrado por microscopia.
• A betatalassemia associa-se a uma ou mais mutações no gene da betaglobina, no cromossomo 11 (em torno de mil mutações diferentes já foram descritas). As alterações genéticas afetam a produção das cadeias de betaglobina que compõem, junto com a alfaglobina, a hemoglobina A, principal componente das hemoglobinas na hemácia. Essa hemoglobinopatia é classificada em três grupos principais: talassemia minor (ou traço talassêmico), intermédia e major. A primeira caracteriza-se por alterações laboratoriais, mas raramente há consequência clínica. Sua identificação, entretanto, é relevante para que seja feito aconselhamento genético adequado. As duas últimas costumam cursar com alterações clínicas de intensidade variável e que envolvem anemia hemolítica com consequente fadiga, deformações ósseas, retardo de crescimento e hepatomegalia, entre outros. Na talassemia intermédia, a produção residual de betaglobina pode amenizar os sintomas, porém indivíduos com talassemia major necessitam de transfusões periódicas de concentrados de hemácias. Quando associada ao gene da Hb S, produz uma variante da doença falciforme chamada de Hb S/betatalassemia.
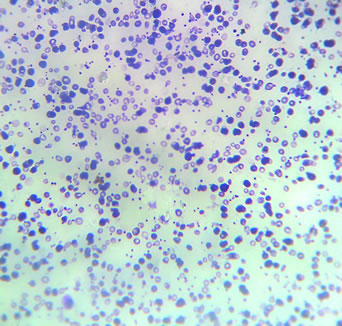
• A alfatalassemia, a seu turno, está associada a mutações no cromossomo 16, que afetam a produção das cadeias de alfaglobina. Indivíduos sem a doença possuem quatro genes de alfaglobina, dois em cada cromossomo. O paciente afetado pode ser portador de mutação ou de deleção em um, dois ou três desses genes. A deleção dos quatro é incompatível com a vida, pois, quando ocorre, causa hidropisia fetal, que leva ao óbito do feto ainda no útero. Os sintomas de anemia são variáveis na alfatatassemia e, de modo geral, relacionam-se ao número de genes acometidos, que, portanto, determina a necessidade de tratamento. Quando o decréscimo da produção de alfaglobina é acentuado, a sintomatologia torna-se mais exuberante. As cadeias beta continuam a ser produzidas de maneira normal e as moléculas excedentes começam a se combinar entre si, originando a Hb H, um tipo instável, que gera quadro de anemia hemolítica e pode se comportar de forma moderada a grave. As consequências clínicas assemelham-se às da betatalassemia. Compare a eletroforese de paciente com Hb Scom a de paciente normal
Compare a eletroforese de paciente com Hb Scom a de paciente normal
Hemoglobina S
Sua frequência varia de acordo com a região do País, podendo chegar a 6%, especialmente em negros e pardos. As formas com manifestações clínicas são conhecidas como doença falciforme e abrangem a anemia falciforme (forma homozigótica) e as combinações com Hb C, betatalassemia ou Hb D.
Hemoglobina C
A prevalência de heterozigotos é da ordem de 1%. Tem maior relevância clinica quando se apresenta em combinação com a Hb S, dando origem a uma forma de doença falciforme. Pacientes homozigotos para Hb C apresentam anemia leve ou moderada, geralmente associada a manifestações clínicas discretas, como cálculo biliar e esplenomegalia.
Hemoglobina D-Los Angeles
Trata-se de um tipo raro de alteração da Hb. Manifesta-se clinicamente quando associada à Hb S (doença falciforme) ou em homozigose, com anemia leve e, ocasionalmente, aumento do tamanho do baço.
Outras variantes
Numerosas outras anormalidades das hemoglobinas e seus genes foram observadas e relatadas no Brasil. Contudo, são exemplos isolados de uma ou poucas famílias portadoras, na grande maioria sem significado clínico.
Como diagnosticar
O diagnóstico das hemoglobinopatias compreende a realização do hemograma, ou análise da série vermelha, além dos testes específicos, que avaliam a quantidade e as características das hemoglobinas. Em situações particulares, a Biologia Molecular pode ajudar o médico a identificar a alteração. Abaixo, estão listados os exames mais úteis nessa investigação.
Eletroforese de hemoglobina em capilar e cromatografia líquida de alta performance (HPLC) Essas metodologias permitem uma detecção visual dos picos de hemoglobina, com seus respectivos valores em porcentagem. Por meio dessa análise, é possível direcionar o diagnóstico, uma vez que um perfil não patológico de hemoglobina possui 97% de Hb A, com 2-3,5% de Hb A2 e 0-2% de Hb fetal. Na betatalassemia minor, ocorre aumento de Hb A2, com níveis em torno de 6%. Já na betatalassemia major, o pico de Hb A está ausente, predominando a fração de Hb fetal. Na presença de picos anômalos de hemoglobina, a junção de duas metodologias torna-se necessária para a identificação correta do tipo de hemoglobinopatia. Contudo, mesmo com o uso dessas ferramentas, nem sempre há possibilidade de chegar ao diagnóstico final. Em tais casos, empregam-se outros métodos.
Outros estudos que podem ser eventualmente realizados:
• Eletroforese em acetato de celulose com pH alcalino e ácido
• Teste de falcização
• Pesquisa de corpos de Heinz
• Pesquisa de inclusões de hemoglobina H
• Pesquisa de hemoglobina instável
• Análise molecular dos genes de cadeias globínicas
Logo após o nascimento
Cerca de 400.000 crianças nascem anualmente com hemoglobinopatias graves. Obrigatório desde a década de 90 no Brasil, o teste do pezinho inclui a triagem para essas doenças e é capaz de identificar a presença de Hb S e outras hemoglobinopatias logo ao nascimento, ajudando a melhorar o acompanhamento e o tratamento dos pacientes acometidos, bem como o aconselhamento genético.
 01 de junho de 2015
01 de junho de 2015Outros destaques dessa edição


Agendar
Exames, vacinas e mais